FDA QSR Project ( FDA 査察対応準備プロジェクト )
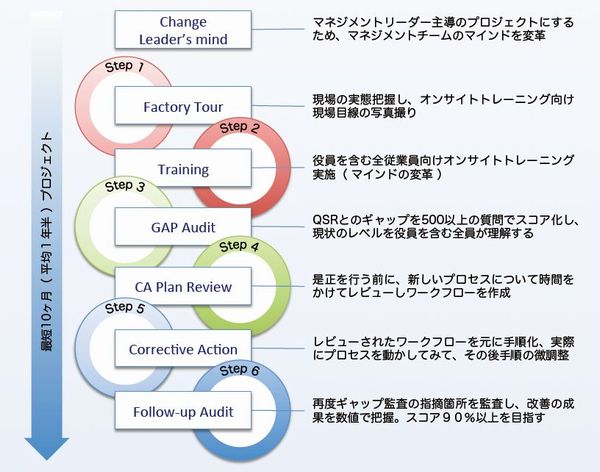
Step0 Change Leader's Mind マインドを変える
グローバルで成功するためには米国市場に於ける成功がかかせません。その為、米国規制に準拠することはが条件となりますが、これはグローバル企業として成功するための条件とも言えます。その結果、大きくフレームワークを変える作業が伴い、大きなプロジェクトで進めていくためのリソースが必要になります。これには、トップマネジメントの理解とリソースの確保がなければプロジェクトは頓挫してしまいます。システムを大きく変更していくためには、トップが旗を振る必要があるのです。
Step1 Fatory Tour 現状の把握
クオリス・イノーバでは、現状を把握するための GAP監査を最初から行っておりません。監査する側とされる側でコミュニケーションが成立せず、両者ともストレスだけが残り監査がまともに終わらないからです。クオリス・イノーバでは必ず GAP監査を実施する前にトレーニングを実施し、グローバル用語の確認から規制の本質の理解を行った上で監査を実施します。さらに、より理解を進めるため、事前にファクトリーツアーを実施し、手順を含む現場の状態を写真に収めトレーニングの際ごご紹介しています。こうすることで理解がすすみます
Step2 Training 本質の理解
3日間のレーニングの内、2日間の QSR基礎トレーニングには、経営陣をはじめ、幹部社員、関連する全ての方にご参会頂き、全員の ISOマインドを一気に医療機器マインドに変革して参ります。全員のマインドの変革無しにプロジェクトの成功は見込めません。このトレーニングセッションでは、国際用語の定義から規制・規格の本質を理解し、GAP監査時のコミュニケーションエラーを排除すると同時に、FDA査察時の誤解による指摘を避けるためでもあります。
Step3 GAP Audit 現実の理解
FDA QSR と ISO QMS とのギャップを確認する監査で、Step2 で学んだ知識を実際の現場で確認し、知識を理解に変えて頂く作業です。また、クオリス・イノーバの550以上に及ぶチェックリストで現状をスコア化(回答率%)し、現状の悪さ加減をマネジメントチームを含む全員に理解して頂きます。これまでの平均は20%に届かないのが現実です。ここまでやって初めて現実を理解することができます。
Step4 CA Plan Review 改善作業
GAP Audit の指摘をプロセス毎にまとめ、全員が何らかの形で参加するプロジェクトチームで改善をしてまいります。クオリス・イノーバでは手順や様式を提供しておりません。他人の手順や様式では自分の物にならず、FDA査察に対応できないばかりか、システムはたちまち崩壊してしまうからです。ただ規制準拠では無く、グローバルに躍進するため、会社にとって真の付加価値となるプロセス作りをサポートしてまいります。
Step5 Corrective Action CAの理解
プロジェクト中にCAPA手順を早めに運用を始め、実際に運用をしながら、CAPAの重要な要素、修正、是正、予防、直接原因、根本原因の相違を理解して頂きます。これらの理解なしに品質改善、システム改善はあり得ず、問題が再発し、品質管理の本質である予防活動が効果的に行われません。これではプロジェクトに意味が無くなってしまうからです。
Step6 Follo-up Audit 理解の確認
GAP監査のスコア(平均20%)が査察対応レベル90%に達したかを確認する為の監査です。モックアップ監査に近い監査となりますが、10ヶ月で90%のスコア達成する為には、かなりリソースをプロジェクトに投入する覚悟が必要です。











