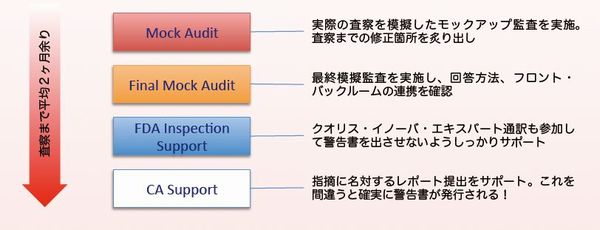
強み1 豊富なFDA査察対応実績
世界各国での豊富なグローバル査察対応実績、警告書を発行させない査察対応プロフェッショナル
| 全世界に100以上の工場を持つ医療機器グローバルリーディングカンパニー・米国本社コーポレート監査スタッフとして、全世界の工場を監査、FDA査察対応実績を持ち、日本企業の査察を支援、パスさせます。( 査察対応プロジェクトでシステムを改善されたお客様に限ります ) |  |
強み2 医療機器・IVDの豊富な知識とノウハウ
長年にわたる臨床現場・開発・製造・RA経験に基づく豊富な知識
| 長年にわたる医療現場、設計、生産技術、RA、品質改善経験を持ち、グロバールフレームワーク、プロセスの豊富な知識と、シックスシグマ、ブラックベルトとしての品質改善ノウハウで、医療機器・IVDの品質システムを一気にグローバルレベルに引き上げます。 |  |
強み3 豊富な経験・実績に裏打ちされたわかりやすいセミナー
1万人以上の受講実績! 具体的事例を交え本質を鋭く説く人気のセミナー
| 臨床現場・医療機器・IVDに関わる豊富な経験に裏打ちされた具体例を元に、分かり易い解説と、患者の命に関わる開発の本質を鋭く説く姿勢が共感と感動を与え、マインドを大きく変革します。オンサイトで行うセミナーでは、経営陣を含む全社員のマインドを一気に変革し、システム改善プロジェクトへの道筋をつけ、査察対応に大きく寄与します。 |  |
強み4 マインドの変革と新たな価値創出
ただFDA査察にパスするだけではなく、全社社員のマインドを変え、新たな付加価値を創出
| FDA査察に対応するだけではく、患者の命を守り、グローバルで成功する会社になるために、全社員をグローバルマインドに変革し、トップマネジメントがリードする全社規模のプロジェクトを通して会社全体の仕組みを大きく変えていきます。グローバルレベルの仕組みに変えていくことで、規制リスクをヘッジし、品質コストを削減し、高利益を生み出すエンジンを作り上げ、付加価値を高めます。 |  |