えっ、どこの認証機関で ISO を取得されたのですか?
ほとんどの日本の会社では ISO を取得されていて、QSR との差分を理解すれば、システムを再構築できると思っておられます。 方向性は間違っていません。 しかし、ここに意外な落とし穴があります!
私達の会社は ISO の認証を取得しているのに、なぜ内容を変更しなければならないのか? それはあなたたの個人的な意見でしょう? 等々。。。 最初どこの会社でも受ける不満の声、声、声 。。 それでは、と、すでにお持ちの ISO のシステムを拝見させて頂くと、どこの認証機関が認証したのですか? と、逆にお聞きしたくなる内容です。 恐らく認証機関のコンサルタントが提供したひな形で作られたのでしょう。 要求事項についての知識が足りないようです。 まったく ISO の知識がない会社さんの方がやりやすいのかもしれません。 そこで、私達はまず FDA QSR の基礎コースのトレーニングを行い、要求されている項目の本質を先に理解して頂くことから始めます。 このことから、ISOが会社にとってあまりプラスになっていないことがおわかりでしょう。 さらに、FDAの要求事項となるとうんざりするはずです。 そこで。。
査察にパスさせるだけならやらない方がいい!
QSRは、Quality System Regulation の略で、米国FDAの医療機器GMPであり、米国に医療機器を輸出する場合、製造業者が準拠しなければならない品質システム規制です。 規制番号は 21 CFR 820。 CFR とは Code of Federal Regulation の略で、アメリカ連邦政府諸機関が施行する50タイトルの規制集の21番目 Food & Drag に区分されている中にあり、その内の医療機器の規制集である第8巻目、820番の番号を付与されている規制がQSRです。 医療機器に関わる規制はこのQSRだけではなく、21CFRの第8巻を見ると、ラベリングの規制があったり、医療機器固有の規制があったりと規制は多岐にわたります。
しかし、重要な要素である品質システムを査察することで、安全で有効性のある医療機器が開発され、製造されることをシステムで保証しようとする考え方が根底にあります。 従って、不具合が発生した場合、CAPAをまわし、根本原因を導き出し、是正した結果を再発防止のために品質システムの手順に落とし込むという改善のための仕組みが要求されます。 規制だから、査察があるから、ではなく、人の命を守るため、品質のよい物を提供しようとする姿勢がないと、やるだけ重荷になるでしょう。 マインドを変えて会社にとってプラスになる方向に向けることがポイントです。
ISO13485との相違についてよく質問があります。 FDAは公にISOと項目は変わらないと言っています。 これはFDAがGHTF( Global Harmonize Task Force )のメンバーであることから対外的に言っていることですが、ISOとは異なる部分が散見されます。 QSRは事実に基づく過去の事例を分析して政府が作った規制であって、産業界の思惑の入ったISOとは異なると考えるべきです。 また、ISOと異なり、QSRを補足説明するガイダンスが多数あるという事実です。 例えば、設計管理について、FDAは設計管理はこうあるべきというガイダンスを出しています。 ガイダンスにはこれに基づく監査はしないと書いていますが、専門家が集まって書いたガイダンス以外の設計管理を査察官に証明するのは至難の業です。 即ち、ガイダンスに従えという事になりますが、このガイダンスはとてもロジカルなもので、考え方を学ぶだけでもプラスになるでしょう。 ISOにはそれがありません。 そして、QSRはアメリカ国民の健康を守る規制、即ち罰則を伴う法律なのです。
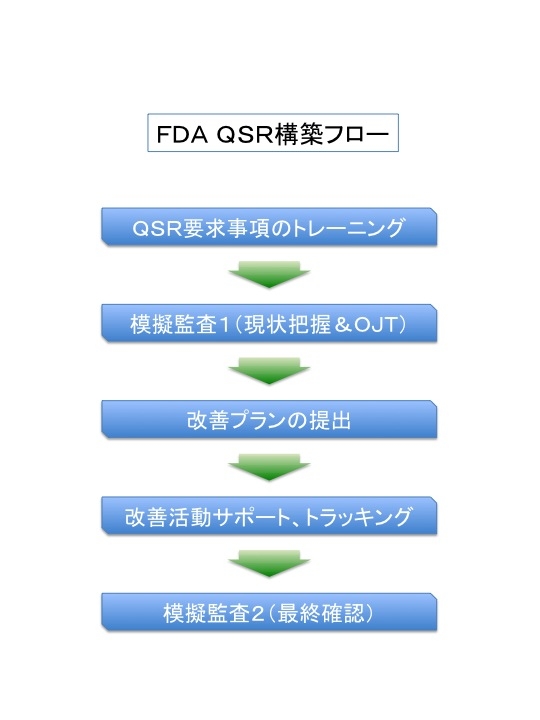
品質システムがすでにあるかないかを問わず、まず、FDA QSRの要求事項のトレーニングを行い、規制の意図する目的を理解して頂き、GAP監査をOJT形式で実施します。 規制の意図していることを実体験して頂くためです。 改善の方向を体験して頂き、提示する改善の方向性を共有して頂きます。 決して負荷となるシステムを構築することが目的ではなく、規制の目的と本質を理解し、”どうせやるなら会社にとってプラスになるシステム作り” をお手伝いします。
クオリス・イノーバは、特に設計管理ガイダンス、CAPAをQSRを理解するために必要なもの、医療機器のみならず製品を開発するための手法として知っておくものという位置づけとして、セミナーのメインコースとしています。
品質不良が査察のリスクを大きくする
機器のリスクレベルが低いから FDA の査察はないだろう。 と思っている会社が多いのも事実。 そこで、なんでそこまでやるの? となってしまいます。 品質不良が発生し、リコールの判断をした場合、当然ながら査察のリスクは高まり、人体に対するリスクがある場合、緊急査察は覚悟しなければなりません。( だったらリコールをしなければいいというコンプライアンスを無視したマネジメントの判断は、市場からの撤退を意味します。 それは、どの国の法律も自国民の健康を守ることが目的だからです。 ) それだけではなく、自社の製品に関係のないところで火の手は上がります。 FDA が最近特に注目している中国製原料の問題は、中国にわざわざ FDA の事務所を開設しなければならないほど事態は深刻になっています。 ( これは、日本への査察の機会が増える可能性も示唆しています。 ) FDA の目線は、製品の開発、製造工程のみならず、品質不良が発生した際のリスクを最小限に留める仕組みと不良の改善への取り組みにも及び、リコール、アクシデントに端を発する査察の場合、当然これらの仕組みが査察されます。( だからCAPAの理解、仕組みを見られるのです ) 品質システムをただの査察対応程度にしか考えていないと、米国市場からの撤退を余儀なくされるでしょう。 リスクをヘッジするためにも品質システムに対するマインドを変える必要がありますが、それにはトップマネジメントのリーダーシップが欠かせません。
Q&A
ISO13485 との違いは?
すでに ISO9000, 13485 を取得しておられると QSR との差分を知りたいとよく尋ねられます。 すでに品質システムが構築されているのだから差分だけ準備すればよいということなのでしょう。 実際には項目別の差分だけで準備をしてもあまり意味がないことに後で気付くことになります。 GAP監査を行うと、現在の品質システムまでことごとく指摘を受けるからです。 私共の QSR Basic Training Course を受講され、 ISO 或いは御社のシステムと何が足りないのか確認していただくのが一番の近道だと思います。 いきなりGAP監査をすると、「 なんでそこまで要求するんだ? 」「 それはあなたの意見でしょ? 」 などとまったくベクトルが合わず、折角のプロジェクトが頓挫することさえあります。 QSRの補足説明資料であるFDAガイダンスは見逃せません。 実はここにISOとの違いがあります。 例えば、設計管理ガイダンスであり、ソフトウェアバリデーションです。
ISO13485 との違いは?(2)
特にマネジメント関連の査察は厳しい質問が矢継ぎ早に続きます。 恐らく ISO のように認証して貰えばよいという考えのシステムでは対応できないでしょう。 QSIT では、マネジメントの査察から始まりますが、どうやって製品の品質を把握し、品質システムの善し悪しを見極めているのかと言ったところが焦点となります。 さらに設計管理は、ISO とは比べものにならない査察となります。 製品品質の善し悪し、不具合の原因のほとんどが設計品質不良であるとFDAは知っているからです。 また、設計、工程管理で共通して深く査察されるのが、検証と妥当性確認です。 これは、ことばの定義から理解していない会社もあるくらいです。 そしてなんと言っても CAPA の指摘が圧倒的に多いのは、恐らく ISO をすでに構築していた会社さんが多いからなのではないでしょうか?
FDAガイダンスとは?
QSRなどの規制の説明や、補助的な役割を持ちます。 例えば、設計管理ガイダンスとは、QSRの設計管理を理解させるために、具体的な例を示して説明をしています。 このガイドラインに従うことで査察をクリアすることができるのです。 しかし、ガイダンスの目的欄には、ほとんど、ガイダンスが規制要求事項ではないと宣言されています。 しかし、ガイダンスは、その道の専門家が集まって作ったもの。 ガイダンス以外の方法で査察官を納得させることはまず無理でしょう。 ISOとの違い、それは、ガイダンスの有無と、規制と同等に扱われることがあるということです。 FDAガイダンスは、本HPリンクページから。
CAPAとは?
CAPA とは Corrective Action & Preventive Action の略で、是正処置及び予防措置のことを言います。 FDAの統計によると、CAPA は指摘の多いものの一つであることがわかります。 FDAの品質システム担当官はこう言いました。 「 CAPA を理解していない会社が多い、特に海外の会社はこれを全く理解していない。」 クオリスではCAPAだけのワークショップコースを設け、実際に CAPA のフォームを作りながら要求事項を身をもって気付いて頂く作業を提供しています。 CAPA を理解して積極的に取り組んでいる会社はあまりありません。 実はこのツールは、シックスシグマにも通じるよいツールです。 本気でやると品質改善もこのツールでできてしまいます。
設計管理とは?
FDA、QSRの設計管理と、ISOの設計管理は何が違うのでしょう? 規制要求事項だけ比べてみると、ほぼ同じように見えます。 しかし、FDAは規制要求事項を理解させるためのガイダンスを発行しています。 設計管理ガイダンスを理解しないと、QSRの設計管理は理解できませんし、ISOの設計管理だけでは、FDAの査察にパスしません。 そして、ガイダンスを読んだだけではピンと来ません。 表面的な説明だけでなく、実践的な例示を行う講習会に参加して理解することをお勧めします。 特にFDA設計管理を習得することをクオリス・イノーバはお勧めしています。 それは、御社のためになる、”設計品質” という宝を獲得するチャンスだからです。

 前のページへ
前のページへ