FDA QSR の要求事項について、要点をまとめています。
弊社 『FDA QSR Training Course』の復習などにご利用ください。
QSRとは「Quality System Regulation」、つまり品質システム規制のことです。
これは、アメリカにおける医療機器に関するGMP(Good Manufacturing Practice、優良製造基準)に該当し、「21 CFR Part820」を医療機器の品質管理における基準として定義しています。
※CFRは「Code of Federal Regulation」、つまり米国連邦政府が定めた規制集を指します。
「21 CFR Part820」には50のタイトルがあります。
医療機器は、この50のタイトルのなかで「Food and Drug(食品と医薬品)」に分類される21番目に区分けされています。その第8巻に医療機器の規制が記載されており、820番がQSRに該当します。このため「21 CFR Part 820」という表記が使われます。
QSRは、アメリカへ医療機器を輸出する製造業者が必ず遵守すべき品質規制です。
この規制へのコンプライアンスを確認するため、FDA(米国食品医薬品局)は外国の製造施設に対しても査察を行います。査察に合格しない場合、Warning Letter(警告書)が発行され、最悪の場合は出荷停止や罰金を科される可能性もあります。
クオリス・イノーバでは定期的に『FDA GMP(QSR)コース』を開催しています。
また、多くの企業でオンサイトセミナーも実施しています。
ご興味のある方は、気軽にお問い合わせください
【参考】
改正薬事法では、国際基準に合わせた整合性のため、GMP(優良製造基準)が『ISO 13485』に基づく形で調整されました。
その結果、多くの企業が『ISO 13485』の認証を取得している可能性があります。
ただし、この認証は法的に義務付けられているわけではありません。
FDA(米国食品医薬品局)は、公には「QSR(品質システム規制)」が『ISO 13485』と整合していると述べています。しかし、その信頼性については慎重に考える必要があります。
FDAのミッションは「公衆衛生を守り、アメリカ国民の健康を保護すること」に重点を置いています。この目的のため、FDAは海外の製造施設にも出向き、監査ではなく正式な査察を実施しているのです。
査察の背景には、アメリカ国民の健康を第一に守るという理念があると言えます。
【参考】
医療機器を海外輸出する企業は、国内販売向けの薬事法監査を受けるほか、毎年、ISO13485やISO9000、14000の監査を受け、さらにCEマークや中国向け規制など、年間を通して監査や査察が続きます。
FDA査察を受ける場合は、会社全体が大騒ぎとなり、通常業務が数カ月間停止する可能性もあります。さらに、毎年の内部監査とそのフォローアップに多くの時間が費やされることになります。
こうした状況を効率的に対応するためには、品質システム専門の組織が必要であることは明白です。
しかし、監査対応にかかる膨大な工数を計算することで、監査対応が企業全体に与える負担が理解できるようになります。工夫次第では、この負担を減らし、コスト削減につなげることも可能です。
FDAのQSR(品質システム規制)は厳しい監査基準を持つことで有名ですが、数多くのガイダンスが提供されています。もともと高い品質を誇る日本製品は、QSRの要求事項を深く学ぶことで、さらなる品質向上と規制遵守を実現できるでしょう。
【参考】
ISO「13485 0.4」は、現在の品質システムをFDAの規制要件に統合することを許容しています。
もしすでにISO「13485」に準拠した品質システムを持っている場合は、足りない箇所を追加して FDA QSRに対応させる必要があります。
統合する際に、「星取り表」を活用して、各手順がISO基準とFDA QSRのどちらに適合しているかを明確にするのが効果的です。このときに重要なのは、情報を文書化して品質システムに組み込み、適合性を示す証拠を準備することです。
ISOの認証を取得した企業のなかには、「うちはISO『9001』とISO『13485』の両方取得しているから大丈夫」と自信を持っているケースをよく見かけます。
しかし、設計手順がないまま認証を取得している場合があります。
驚くことに、これは特定の企業の話というわけではありません。ISO『13485』の人気が高いため、監査員が不足してしまい、得意分野だけを確認している監査員が増えていることが一因とされています。
また、アメリカのFDA査察官は認証機関(Notified Body)が発行する認証書にあまり関心を持ちません。
アメリカではISO基準の馴染みが薄いというだけではなく、そもそも他国の規制にも関心が低い傾向があります。そのため、「『ISO13485』を取得しているからFDAの監査をパスできる」という考えは危険であり、実際にはパスすることが非常に難しいのが現状です。
【参考】
査察でよく使われる用語は、日常的に社内でも使い慣れておくと便利です。査察時に慌てないようにしましょう。
特に以下の3つは必ず覚えておきましょう:
重要なポイントは、これらに該当する社内文書をリストに明記し、適切に管理することです。
「V&V」とは、Verification(検証)と Validation(妥当性確認)を指します。
「検証」と「妥当性確認」を混同していたり、意味を正しく理解せずに使われているケースをよくみかけます。
設計管理や工程管理で重要なポイントになるので、規程や手順書内で明確に定義しておくことが大切です。実際、FDAの査察時に設計管理でもっとも多く指摘される項目です。
クオリス・イノーバでは、設計管理ガイダンスコースを設け、これらの概念を丁寧に解説しています。
経営者の責任として次のようなことが挙げられます。
内部品質監査の目的は、QSRの要求事項への適合性を確認し、品質システムの有効性を評価することです。
指摘があった場合は、是正処置を実施する必要がありますが、この是正処置を正確に理解し、根本原因が解消された企業はほとんどありません。
さらに、有効性を示すべき報告が、単なる指摘件数の羅列になっている場合があります。
こうした背景には、ISOの運用が適切に行われていない現状があり、その主な原因は内部監査の不備です。加えて、第三者認証機関がその不備を追認する報告書を提出していることが、問題をさらに深刻化させています。
このような「負のスパイラル」を断ち切る必要があります。
要員については、必要な教育、経歴、訓練、および経験を備えた人材を雇用することが求められています。
社員から「どの規程やSOP(標準作業手順書)を知っておくべきか」と質問された際、明確に回答できなければ、監査で指摘される可能性があります。
実際に、是正処置(CAPA)を実施するなかで、根本原因の多くが「手順を知らない」「十分な教育を受けていない」ことに起因していることがわかります。つまり、必要な教育や知識の提供を徹底することが重要だと言えます。
安全性や有効性を含んだ医療機器の品質は設計段階で決まります。
設計管理の手順では、開発計画から製造部門への設計移管まで、QSRが要求する基準に従った手順を設定する必要があります。FDAの設計ガイドラインでは、ひとつの概念図を用いて設計管理を説明しており、この図を通じて開発の各ステップにおけるデザインレビューの重要性や、検証と妥当性確認の関係性を明確にしています。
なお、クオリス・イノーバが提供している「設計管理ガイダンスコース」では、医療機器開発の本質をお伝えしています。
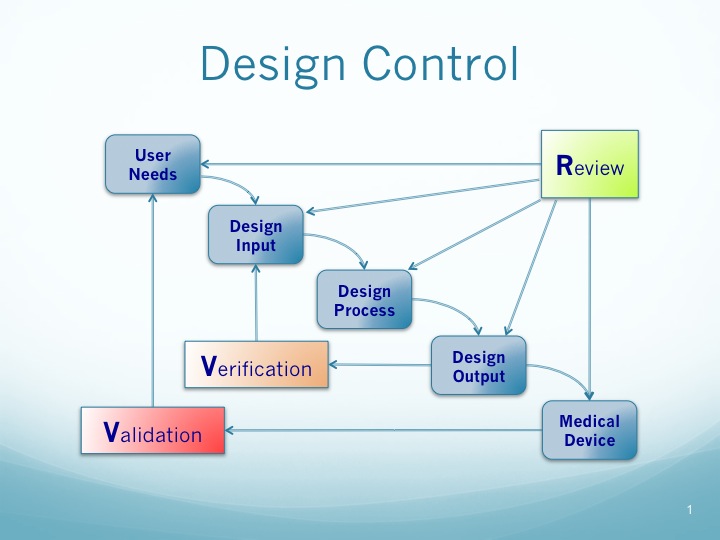
製品開発を進める際には、設計計画書を作成します。
この計画書には、進捗を管理するためのガントチャート、必要なスキルを持った担当者の配置、そして開発にかかる費用の管理などが含まれます。
設計計画書では、プロジェクトマネジャー、リーダー、そして各部門を代表するメンバーを明確にし、設計レビューのどの段階で誰が関わるかを事前に定めておくことが重要です。
また、この計画書は変更が加えられる場合があるため、リビジョン管理が適切に行われていることが求められます。
監査員の視点では、設計計画書を確認することで、製品設計が適切に管理されているかどうかを判断することができます。
設計のインプットとは、ユーザーや患者の漠然としたニーズを具体的に数値化した仕様に変換するプロセスです。
たとえば、ユーザーが「持ちやすい長さ」と言った場合、それを「10 cm ± 2 mm」という具体的な長さを導き出す必要があります。さらに、その手順と判断を裏付ける証拠が求められます。
この重要な作業を怠ると、製品完成後のバリデーション(意図した用途の確認)でユーザーから否定される可能性があり、設計をやり直すことになりかねません。これにより開発コストが増加するリスクがあります。
監査員は「どうしてこの設計値になったのか?」というシンプルな質問を通じて確認を行います。
FDAは、この設計インプットを「製品開発期間の約30%を費やしてもいい」としているほど、重要視しています。
なお、クオリス・イノーバが提供する「設計管理ガイダンスコース」では、トレーサビリティーマトリックスの使い方について詳しく説明しています。
「設計アウトプット」とは簡単に言えば、図面、検証結果の技術レポート、製造時に使う基準書などのことです。
まずは、どの書類が設計アウトプットにあたるのかをきちんと定義しておきましょう。
設計インプット(設計の要求事項や開発スペック)が決まり、そこに許容差なども盛り込まれると、その内容に基づいて検証のルールや試験方法、合否判定の基準をあらかじめ設計計画の中に盛り込みます。
※ きちんとした「計画」がなければ、正しい検証はできません。
実際には、「設計インプット」と「設計アウトプット」をしっかり計画した時点で、設計作業の約8割は完了しているとも言えます。その後は、試作品を作り、動作を確認し、計画通りの試験を行って、スペック(要求仕様)を満たすまで、何度か改良を繰り返します。
合格した試験結果(検証記録)ができあがれば、それを元にした図面とあわせて、それらが正式な「設計アウトプット」として扱われます。
監査の際には、「設計インプットと設計アウトプットがどう結びついているか」が重要な確認ポイントとなります。
文書管理はFDA監査において非常に重要なポイントです。
FDAが特定の手順を要求している場合、その手順は必ず整備されていなければなりません。
この手順では、関係者のニーズを把握し、必要な教育を行い、その結果として力量(習熟度)を確認します。
文書管理が必要な範囲はとても広いため、本来管理すべき部門がそれを十分に行えていなかったり、ISOでは当然のことがきちんと守られていないケースも少なくありません。
FDAの査察は、多くが書類ベースの確認(机上監査)で進められます。
そのため、提出された文書から組織の管理状況や運用実態など、非常に多くの情報を読み取ることができます。
日本の企業では、昔からサインの代わりに名前入りの日付印を押す習慣があります。これは日本や韓国、中国など漢字文化圏に多く見られます。実は日本の産業界が効率化の名目で広めた文化でもあります。
一方、アメリカではサインそのものが自己責任を伴う重要な行為とされています。
そのため、FDAの査察官の目には、誰でも押せる日付印を使って承認している行為が非常に奇妙に映っています。大企業でも、この日付印が悪用され、金銭トラブルにつながるケースもあります。
また、FDA査察官は、個人の印鑑や三文判を承認に使う行為も大きな問題と考えています。
特に、印鑑が机の上に放置されていたり、施錠されていない場所に保管されていたりする状況は理解されません。
結果として、文書を適切な責任者がきちんと確認・承認した証拠がないと判断されかねません。査察案は、「代わりに誰かが押印している可能性がある」と結論づけてしまうからです。
もしFDA査察を受けるなら、印鑑ではなくサイン方式へ切り替えることが不可欠です。抵抗はあるかもしれませんが、このままではFDA査察に合格するのは難しいでしょう。
購買部門の主な目的は、品質が高くコストが抑えられる製品やサービスを提供する業者(サプライヤー)を評価・選定することです。規制では、業者を選定するための基準を設け、品質要求事項を満たす業者を選ぶことが求められています。
査察のポイントは以下の3つです:
識別管理の適用範囲に注意しましょう。
識別管理の適用範囲は広く、製品が工場で受領されてから出荷、流通、据付されるまで、一貫して管理することが求められています。 つまり、工場内のすべての製品がしっかりと識別管理されている必要があります。
特に注意すべきは、不適合品や試験用サンプルです。
たとえば、技術部門やマーケティング部門が使うサンプル品などが、識別されないまま倉庫に無造作に置かれているケースがよく見られます。
また、完成品については、「出荷されるまで隔離する」ことが**820.80(d)**で求められています。 ここでいう「隔離」とは、出荷責任者のサイン(承認)が完了するまでは出荷できない仕組みを意味しています。
監査員の視点では、たとえば良品と不適合品が、単にラインやトラテープ(床の仕切り)だけで隣に置かれている場合は指摘対象になります。
欧米では、不適合品は「鍵付きケージに入れて隔離する」ほど厳しく管理されているのが一般的であり、日本とは文化が違います。 こうした文化の違いを理解することが重要です。
この規定は内容が非常に多く、これだけでセミナーが開けるほど重要なテーマです。
また、すでに設計が完了している段階のため、査察官から特に指摘を受けやすい部分でもあります。
経験豊富な査察官であれば、工場内を一通り確認した後、すぐに書類審査に移るでしょう。
査察時の主なチェックポイントは次の通りです:
これらのポイントについては、QC工程表(品質管理工程表)をもとに各工程ごとに確認し、その内容を記録として残すことが重要です。
査官が注目する項目は、設計移管、設計変更、工程変更、要員の適性評価、そしてCAPA(是正措置および予防措置)の実施状況です。
生産ラインで使用する装置は、測定すべきスペックを正確に満たせるものである必要があります。
たとえば、スペックが「3.5V ± 0.005V」であれば、小数点3桁目まで測定可能なデジタルボルトメーターが求められます。しかし、現場では小数点2桁目までしか測定できない装置が使用されるケースが多く見られます。
また、校正期限が過ぎている測定器や、校正管理されていない測定器が存在する場合もあります。
こうしたことは本来、**工程の検証(バリデーション)**の段階で確認・整備しておくべきものです。
また意外と見落とされがちなのが、日々の点検です。
せっかく点検シートを用意していても、きちんと活用されていないケースがしばしば見受けられます。
こうした状況を防ぐには、週1回の工程点検(ウィークリー点検)などを取り入れて、現場に適度な緊張感を持たせる工夫が必要でしょう。
製造工程で行われる試験は、製品の品質を直接保証するものではなく、製造手順が適切に実施されたことを確認するための試験に過ぎません。
しかし、本来ものづくりで目指すべき理想は「試験のいらない品質保証(無試験検査)」です。 そのためには、製品の品質に影響を与える変動要因(パラメータ)を正しく理解し、製造中にそのパラメータを常に監視する体制が必要です。
たとえば、溶着工程で製品をランダムに選んで破壊試験を行う方法では、試験に手間やコストがかかるうえ、試験されなかった製品に不良が混ざるリスクも常に存在します。
このようなムダやリスクを避けるには、さまざまな要因を組み合わせて試験を行い、どの因子が品質にもっとも大きく影響するかを明らかにして、重要なパラメータを継続的に監視することが求められます。
これを実現するには、明確なプロセスバリデーション手順が必要です。
バリデーションの本質を理解し、VMP(バリデーションマスタープラン)、DQ(設計の妥当性確認)、IQ(設置の妥当性確認)、OQ(運用の妥当性確認)、PQ(性能の妥当性確認)の各ステップがなぜ重要なのかをしっかり学ぶことが大切です。
クオリス・イノーバの「プロセスバリデーションコース」は、こうした本質を実践的に学べるセミナーです。
Subpart H(Acceptance Activity)は、「受け入れ検査」と誤解されやすいため、原文のまま「アクセプタンス活動」と表記されています。
このアクセプタンス活動には、「受入」「工程内」「完成機器」の3つの段階があります。
なかでも Final Acceptance Activities(最終出荷承認)は規制が非常に厳しく、出荷承認が完了していない製品については誤出荷を防ぐために隔離管理が義務付けられています(ここは特に注意が必要です)。
査察官は、この承認活動を通じて企業の品質管理状況をさまざまな角度から確認しています。
そのため、「逸脱」「特別採用」「修正」「修理」などの用語の定義やその理解を正確にしておくことが求められます。
生産工程で不適合品(Nonconforming Product)が発生した場合、FDAの査察官は「なぜ不適合品が発生したのか」「原因をきちんと分析しているのか」を必ず確認してきます。
なぜなら、妥当性確認(バリデーション)が済んでいるはずの工程で不良品が出ているということは、その妥当性確認自体に問題があった可能性があると判断するからです。
不適合品が発生した際には、是正措置および予防措置(CAPA)を適切に実施する必要があります。しかし実際には、CAPAの本質を正しく理解し、効果的に実施している企業はまだ少ないのが現状です。
査察官、特に経験豊富な査察官(エキスパート)は、まず最初に不適合品の処理レポートを確認します。このレポートを見ることで、その企業の品質管理の実態がよく分かるからです。
いわゆる「CAPA(是正および予防処置)」は、FDA査察官が特に重視するポイントです。
重要なのは、規制で求められているすべての入力(インプット)に対してCAPAを実施することです。(実際にはこの部分で多くの指摘を受けています) そして、CAPAの7つのステップを確実に実行することが求められます。
しかし、実際に企業のCAPAシートを確認してみると、多くの場合それは「修正」にとどまっており、正しい意味での「是正処置」になっていません。ISOでの品質改善のやり方に慣れている方は、これに気付かないことが多く、指摘してもなかなか理解されません。その結果、同じ品質問題が繰り返されてしまいます。
根本原因が正しく特定されず、その場しのぎの対処(モグラ叩き)だけでは、潜在的な原因を統計的に分析・解決することができず、当然再発リスクは高まります。
さらに、日本のメーカーの多くは「CA(是正処置)」と「PA(予防処置)」の違いを理解しておらず、監査の際にPAのデータを提出できないケースが目立ちます。
クオリス・イノーバがCAPAだけをテーマにしたセミナーを開いている理由は、この分野こそがFDAも私たちも重視する「品質改善」と「コンプライアンス」の基盤だからです。実際、FDAの指摘事項の中でもCAPAに関するものがもっとも多くなっています。
ISOの感覚のままではFDA査察をクリアできないことに、早めに気付いていただきたいと思います。そして、単に規制遵守(QSR対応)だけでなく、品質改善がいかに企業の利益や機会損失の防止につながるかという視点を持つことが大切です。それこそが、私たちのセミナーで伝えたい最も重要なメッセージなのです。
「ラベリング」という言葉には、単なる製品ラベルだけでなく、添付文書や取扱説明書などもすべて含まれることに注意が必要です。
QSR(品質システム規制)では、ラベリングに関してDHR(Device History Record:製造履歴記録)への記録と、検査担当者の署名を求めています。これは、ほかの項目にはあまり見られない表現です。
FDA査察では、「ラベルの取り違え」や「誤情報の混入」などを防ぐために、企業が混同をどう防止しているかが重要なチェックポイントになっています。実際、ラベリング関連の不備はリコールの原因としてもっとも多いため、FDAの審査基準が非常に厳しくなっています。
FDAの査察では、医療機器の包装設計図や輸送用の外箱の設計図、そしてそれに関する検証レポートの提示が求められます。これは、機器が保管や流通の過程でしっかり保護されるように設計されているかを確認するためです。
そのため、実際の輸送状況を想定した試験や、シミュレーションに基づく検証データを用意しておく必要があります。
また、設計だけではリスクを完全に防げない場合、外箱に「取り扱い注意」や「積み重ね禁止」などの注意表示やマークを付けることで対応します。たとえば「箱は6段までしか積んではいけない」と定めた場合、その制限の根拠となる試験結果や設計情報を用意しておく必要があります。
この項目では「製品の取り扱い」に関する規制について説明されています。
ISOの定義では“製品”に中間製造品(途中段階の部品や部材)も含まれるので注意が必要です。つまり、修理や保守に使うサービスパーツも“製品”と製品と見なされるこということです。
また、製品の汚染や劣化には特に注意が求められます。
たとえば、木枠に梱包された製品が晴れた日ににトラックから降ろされ、製造所に入る順番待ちで長時間日光にさらされるケースがあります。査察時には雨天になることもあり、そうした環境管理の問題が指摘される場合があります。
そしてもっと問題なのは、査察官がその日の監査を終えて帰るときにも、まだその製品が屋外に置かれているような状況です。こうなると、「製品管理が適切に行われていない」と見なされるリスクがあります。
ここで求められているのは、倉庫などでの製品の「混同」「損傷」「劣化」やその他の悪影響を防ぐことです。そのため、それらを防止するための具体的な手順が整備されているかが査察で確認されます。
出荷前の製品倉庫を運送会社にアウトソーシングするケースも多いですが、たとえアウトソーシングしていても、最終的な責任は製造業者にあることを忘れてはいけません。
アメリカでは、倉庫内のシロアリや害虫の防除対策は当然のこととされており、定期的なメンテナンスも実施しています。
また、FDAの査察官は、梱包箱に印刷されているマーク(取扱注意や積み重ね禁止など)にも注目します。そこに製品管理の適切さを判断する多くのヒントがあるからです。
出荷承認されていない製品が市場に出回らない仕組みがあるかどうかが査察で問われます。さらに、有効期限が切れた製品が流通しないように管理されているか、そのための標準作業手順書(SOP)の整備も必要です。
また、製品がどのように流通したかを示す記録(伝票など)も必要ですが、その伝票に法規制で求められている情報がきちんと記載されているかどうかを必ず確認してください。
インスタレーションマニュアル(設置手順書)は、実際に正しく設置が行われるように、検証やバリデーション(妥当性確認)を実施する必要があります。
設置作業は、ある意味で製造工程の延長であり、製品の性能に大きく影響する重要な工程と考えられます。そのため、適切な治具や測定機器を使って、正確に作業が行われることを事前に確認しておくことが大切です。
記録は「客観的な証拠(Objective Evidence)」として非常に重要です。
口頭での説明は基本的に受け入れられず、必ず記録で証明する必要があります。そのため、必要な情報が記録に残るよう、手順を見直すことが求められます。
特にV&V(検証と妥当性確認)の記録が不足している場合が多く、査察官が重点的にチェックするポイントとなるため注意が必要です。
査察準備をする際には、査察官と直接対応する部屋とは別にバックルームを設け、必要な書類をすべて整えておき、査察官からの要求に迅速に応えられる体制を整えることが求められます。査察官から要求された書類をすぐに提示できないと、それだけで指摘対象になる可能性があるため、注意が必要です。
DMR(Device Master Record)とは「機器原簿」のことです。
これは、医療機器を製造するために必要なすべての文書をまとめたもので、日本の薬事法でいう「製品標準書」に相当します。
具体的には、機器の仕様書、インスタレーションマニュアル、修理手順書などもDMRに含まれます。そのため、自社のどの文書がDMRに該当するのかを明確に定義した文書を用意しておくことが重要です。
FDAの査察では、設計変更に応じてDMRの内容もきちんと更新されているかがチェックされます。記録の一貫性と正確な管理が問われるポイントです。
DHR(Device History Record)とは、「製造記録」のことです。
これはFDAの査察で必ず確認される重要な文書で、各製品が設計通りに製造されたことを証明するための記録です。
注意すべき点は、DHRに日付印(印鑑など)を押すことによる指摘のリスクが高いことです。印鑑を押すという行為は、事前に印刷された名前と同じとみなされ、信頼性に欠けるとして指摘される可能性があります。そのため、署名(サイン)を使う方が安全です。
また、DHRには使用した測定器がトレースできるように記録しておくことが求められます。
さらに、不適合が製造ラインで発生した場合、どのように対応したかの記録も必要です。もし最終的に「適合」と判断された場合には、**その判断に至った根拠となる証拠(エビデンス)**がファイルに残されていなければなりません。
熟練した査察官は、DHRを見ただけでさまざまなことを読み取ります。
たとえば「なぜこの図面のリビジョンが変更されたのか?」「この作業員のスキルは?」「トレーニングは受けているか?」「DMRとの整合性はどうか?」など、細かな点まで見抜かれると考えておきましょう。
品質システム記録とは、特定の製品ではなく、QSRが求める手順や文書、特に820.20(経営者の責任)で指定された記録を指します。
そのため、どの記録が「品質システム記録」にあたるのか、文書番号などを明記して定義しておくと、社内での混乱を防ぐことができます。
まず理解しておくべきなのは、FDA(アメリカ食品医薬品局)の最大の使命は、アメリカ国民の健康と安全を守ることだという点です。このため、アメリカから寄せられる苦情が迅速かつ適切に処理されているかどうかが、査察官の重要なチェックポイントになります。
苦情の内容によっては、**MDR(Medical Device Reporting:医療機器不具合報告)の対象となる場合もあります。MDRは別の規制「Part 803」**で具体的な報告義務が定められており、MDRに該当するかどうかを正しく判断し、その対応ができていなければ、査察で指摘を受ける可能性が高くなります。
また、苦情に関する調査記録は詳細な要件が定められており、ここも指摘の多いポイントです。つまり、FDAの査察官が特に重視する領域であるということを意識して、万全な準備をしておく必要があります。
付帯サービス(820.200)の(b)では、サービス記録の分析を行い、必要に応じてCAPA(是正措置・予防措置)のプロセスを適用することが求められています。
また、その分析結果や対応状況は、マネジメントレビュー(経営者レビュー)で報告されなければなりません。さらに、MDR(不具合報告)に該当するかどうかを判断するためのプロセスも整備する必要があります。
統計的手法の適用範囲は、手順とともにISOであらかじめ定められています。
QSRでは、この統計的手法を工程能力の検証と製品特性に活用することが求められています。もちろんほかの場面でも統計手法は有効ですが、少なくともこの2つの用途には確実に活用できるようにしておく必要があります。