| ! | 本コース受講前に、QMSR2日間コースの修了を強くお勧めします。設計管理はQMSの一部であり、システム全体の理解が学習効果を大きく左右します。 |
なぜ今、設計管理が最重要課題なのか
日本の医療機器メーカーの多くは、設計・開発プロセスが世界基準に対して大きく遅れているという厳しい現実があります。クオリス・イノーバが20年以上にわたって積み重ねてきた監査データは、医療機器の品質問題の根本原因が「製品の設計段階」にあることを統計的に証明していて、これはFDAの査察においても繰り返し指摘される事実です。Form 483の件数分析からも、設計管理の不備が最多の指摘項目であることが明らかです。設計の失敗はリコール、製造停止、そして最悪の場合、患者の命に関わる事態を引き起こします。
このコースで得られる3つの本質的な価値 - 「規制だからやる」を超える — 設計品質向上が患者の命を守り、企業競争力を高める理念を体得する
- グローバル開発フレームワークの習得 — FDAガイダンス・ISO 13485・QMSRに準拠した設計管理を体系的に理解する
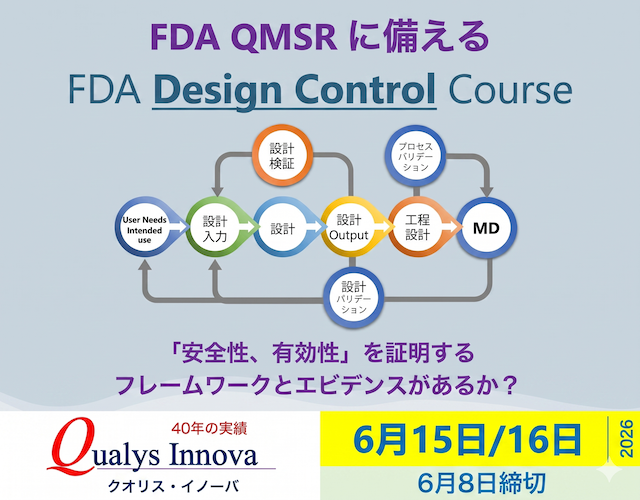
- 工程設計まで一貫して学ぶ — 製品設計から製造移管・プロセスバリデーションまで、2日間で全体像を把握する
|
対象者
- 医療機器・IVD市場へこれから参入しようとしている企業のエンジニア・品証担当者
- 海外(米国・EU等)への輸出・進出を計画している企業の開発・薬事担当者
- FDA査察・MDSAP受審を準備している企業のプロジェクトチーム
- 既にFDA査察を終え、Form 483・Warning Letter指摘事項への対応に苦慮している企業
- 製品実現に関わるすべての部門(マーケティング・開発・生産技術・品証など)
コース概要
本コースは、FDA「設計管理ガイダンス(Design Controls Guidance)」、ISO 13485、および新規制QMSR(Quality Management System Regulation)をもとに、グローバルな医療機器開発と設計管理の方法論を体系的に解説する2日間の集中プログラムです。単なる規制解説にとどまらず、タグチメソッドを用いたロバスト設計、ライフサイクルリスクマネジメント(LCRM)、コンカレントエンジニアリング、フロントローディング手法など、エンジニアリングの本質に根差した実践的アプローチを習得できます。2日目は設計移管後の工程設計とプロセスバリデーション(VMP/DQIQ/OQ/PQ/PPQ)を詳細に解説し、「検査に頼らない品質保証」の実現方法を学びます。
◆開催日程◆
- 日 時:2026年 6月15日(月)、16日(火) 9:30〜16:30
- 締 切:2026年 6月8日
- 会 場:ハイブリッド開催
会 場:全国町村会館【東京・永田町】
リモート:ZOOMのみ - 講 師:クオリス・イノーバ株式会社 代表取締役社長 木村 浩実
- 定 員:会 場:12名
リモート:60名 - 受講料:99,000円(テキスト)会場のみ昼食あり
88,000円(2名様以上ご出席)
継続研修をご希望の方はご相談下さい - 修了書:FDA査察時にエビデンスとして使用可能
- お申込:セミナー申込み
リモート受講の方 こちらから
会場で受講の方 こちらから |
◆セミナー内容◆
弊社発行、「FDA 21CFR Part 820 QMSR Ver.2」、「FDA Inspection of Medical Device Manufacturers CP7382.850」をご持参ください。
| A|コンプライアンスとインテグリティー | 01 | 設計不良の根本原因 | ケーススタディ分析 / 設計CAPA / 不正排除の仕組み | | 02 | 規制・行政のミッション | 「規制適合」で十分か? / 患者の命と企業倫理 / FDAミッションとの整合 | B|医療機器規制の最新動向 | 03 | QMSR最新対応 | 新規則QMSRの全体像 / 既存QSRとの差異 / 移行計画の考え方 | | 04 | 適用規制・規格体系 | 21 CFR Part 820 / ISO 13485 / IEC 62304 / ISO 14971 | C|製品実現プロセス(ISO 13485 第7章) | 05 | 製品実現の全体構造 | LCM(ライフサイクルマネジメント) / 安全性と有効性の立証 / 顧客フィードバック活用 | D|設計管理 ISO 13485-7.3 – フレームワークと逐条 | 06 | (a) 総則・プロセスアプローチ | タートルチャート / 力量管理820.25 / 文書体系の俯瞰 | | 07 | (b) 設計・開発計画 | LCM / LCRM / コンカレントエンジニアリング / タグチメソッド・フロントローディング | | 08 | (c) 設計インプット | ニーズの明確化 / トレーサビリティーマトリックス(TMX) / ラベリング・包装設計 | | 09 | (d) 設計アウトプット | E-BOMの役割 / 製造仕様書 / タートルチャートのアウトプット | | 10 | (e) 設計レビュー | 早期問題発見の仕組み / テクニカルレビューとの違い / レビューワーの責任と権限 | | 11 | (f) 設計検証 | インプット〜アウトプットのトレーサビリティー / エビデンスの作り方 / 検証計画と検証レポート | | 12 | (g) 設計バリデーション | 検証とバリデーションの相違 / ソフトウェアVモデル / 3種類のバリデーション | | 13 | (i) 設計変更 | 変更管理プロセス / LCRMとの連動 / 変更フロー | | 14 | (j) DHF | DHF / DMR / DHR の定義 / 文書体系と設計標準 / ファイル管理の実務 | | 2日目 設計移管と工程設計・プロセスバリデーション | E|設計移管( ISO 13485-7.3.8) | 15 | 設計移管の本質 | LCRMにおける設計移管 / 均一製造を実現する手法 / プロセスバリデーションの目的 | F|プロセスバリデーション(ISO 13485-7.5.6) | 16 | なぜ工程バリデーションが必要か | 本質と目的 / 検査依存からの脱却 / 規制・規格の要求事項 | | 17 | バリデーション対象工程の識別 | 検証工程とバリデーション工程の相違 / 設備バリデーションとの違い / バリデーション工程例 | | 18 | バリデーションの種類と計画 | VMP:バリデーションマスタープラン / DQ / IQ / OQ / PQ / PPQ / 文書化プロセス | | 19 | CSV(コンピューターシステムバリデーション) | CSVカテゴリー / リスクベースCSV / ソフトウェアバリデーションの基本 | G|工程設計とプロセスリスク分析 | 20 | 工程設計の進め方 | PFMEA(工程FMEA) / QC工程表・PTMX / 環境・汚染・副資材管理 | | 21 | バリデーションのアウトプット | 工程能力(統計的手法) / リバリデーション設定 / 設備インスペクションとメンテナンス | | 22 | 回顧的バリデーション | 適用条件と考え方 / 既存工程への対応 / 記録の整備方法 |
|